Emerging Variant Glioma: Glioblastoma with a Primitive Neuro-Ectodermal Tumor(PNET) Component
Article information
Abstract
A 81-year old male with multiple mass lesion of the right fronto-temporo-parietal lobe and right frontal lobe was admitted to our hospital due to hemiparesis. Neuroimaging studies showed ring-enhancing lesions in right fronto-temporo-parietal lobe and right frontal lobe on magnetic resonance imaging (MRI). Tumor tissue evaluated to be consistent with glioblastoma radiologically was subjected to total excision. Using a morphological and immunophenotypic approach, the predominant component of the tumor was found to bear the properties of classic glioblastoma. The other component was composed of undifferentiated areas that exhibiting small cell morphology and diffuse neuronal immunophenotype. The case was diagnosed as ‘Glioblastoma with primitive neuroectodermal tumor (PNET) component’. The patient who had been performed postoperative radiotherapy, showed shrinkage of non-operative lesion and no sign of recurrence and neurological deficit during 12 months follow-up. We report a rare case of glioblastoma with a PNET component that has a relatively favorable outcome following surgery with radiotherapy and then review previous reports.
INTRODUCTION
Glioblastoma is the most common primary brain tumor in adults, and exhibits a heterogeneous histological spectrum2,10). Marked diversity exists in the clinicopathological characteristics of glioblastoma and recent studies have suggested the presence of heterogeneity provides an indication of the disease’s nature1,3,8,9). The most recent guidelines for brain tumors suggest there is sufficient evidence to support the presence of classic glioblastoma, gliosarcoma, and giant cell glioblastoma. Furthermore, the guidelines have suggested the possibility of various emerging glioblastoma variants10).
In emerging glioblastoma variants, primitive neuroectodermal features have been reported as a potential variant of glioblastoma and have been present in older adults7). Primitive neuroectodermal tumor (PNET), is an aggressive neoplasm encountered more commonly in the pediatric population. Defined as a primitive, it is expected to demonstrate potential for divergent differentiation along glial, neuronal cell types11).
It is rare to encounter a brain tumor, demonstrating or presenting both primitive neuronal cells and elements of more advanced glial cells, specifically astrocytic differentiation15). As in glioblastoma, the prognosis for central nervous system PNET is poor, although the response to therapy is more frequent and long-term survival is somewhat better for PNET5).
We report a rare case of glioblastoma with a PNET component that has a relatively favorable outcome following surgery with radiotherapy and then review previous reports.
CASE REPORT
An 81-year old male presented with a three day history of left hemiparesis and a sudden onset of left facial paresis. A neurological examination showed an alert mentality with a Glasgow Coma Scale of 14 and revealed left hemiparesis (motor Grade IV) with central type left facial palsy. Brain magnetic resonance imaging (MRI) revealed a space-occupying lesion in right fronto-temporo-parietal lobe. This tumor demonstrated a heterogeneous high signal on T2-weighted images and an iso-low signal on T1-weighted images with signal voids suspected to be a vascular structure (Fig. 1).

Pre-operative evaluation MRI (Axial T1-WI MRI with enhancement): Illustrates each separated heterogeneous hypointense intra-axial lesion in the right frontotemporal lobe (4.5×4.0×4.0 cm, A) and right frontal lobe (2.5×2.5×2.0 cm, B), areas of cystic formation and infiltrative lesions with significant surrounding edema and mass effect. Image shows heterogeneous masses with an irregular ring of enhancement and central necrosis with surrounding signal abnormality, which is probably associated with tumor extension and edema.
A whole body positron emission tomography demonstrated hypometabolism in the right hemisphere without metastatic lesions from another organ. Based on the clinical and radiological findings, we planned to perform a surgical excision for decompression and pathologic diagnosis. In the operative field view, the tumor’s gross appearance was a dark and hard consistency within a surrounding adhesion on the normal brain parenchyme.
Histologically, the predominant component of the tumor was found to bear the properties of glioblastoma. Microvascular proliferation, mitotic activity, as well as geographic and pseudo-palisading necrosis, were present. The other component was composed of undifferentiated areas that exhibiting small cell morphology and diffuse neuronal immunopheno-type (Fig. 3). Immunohistochemistry showed the tumor cells were negative for CD20, CD79a, CD3, and EBV. Some tumor cells were positive for GFAP, CD56, and Ki-67.
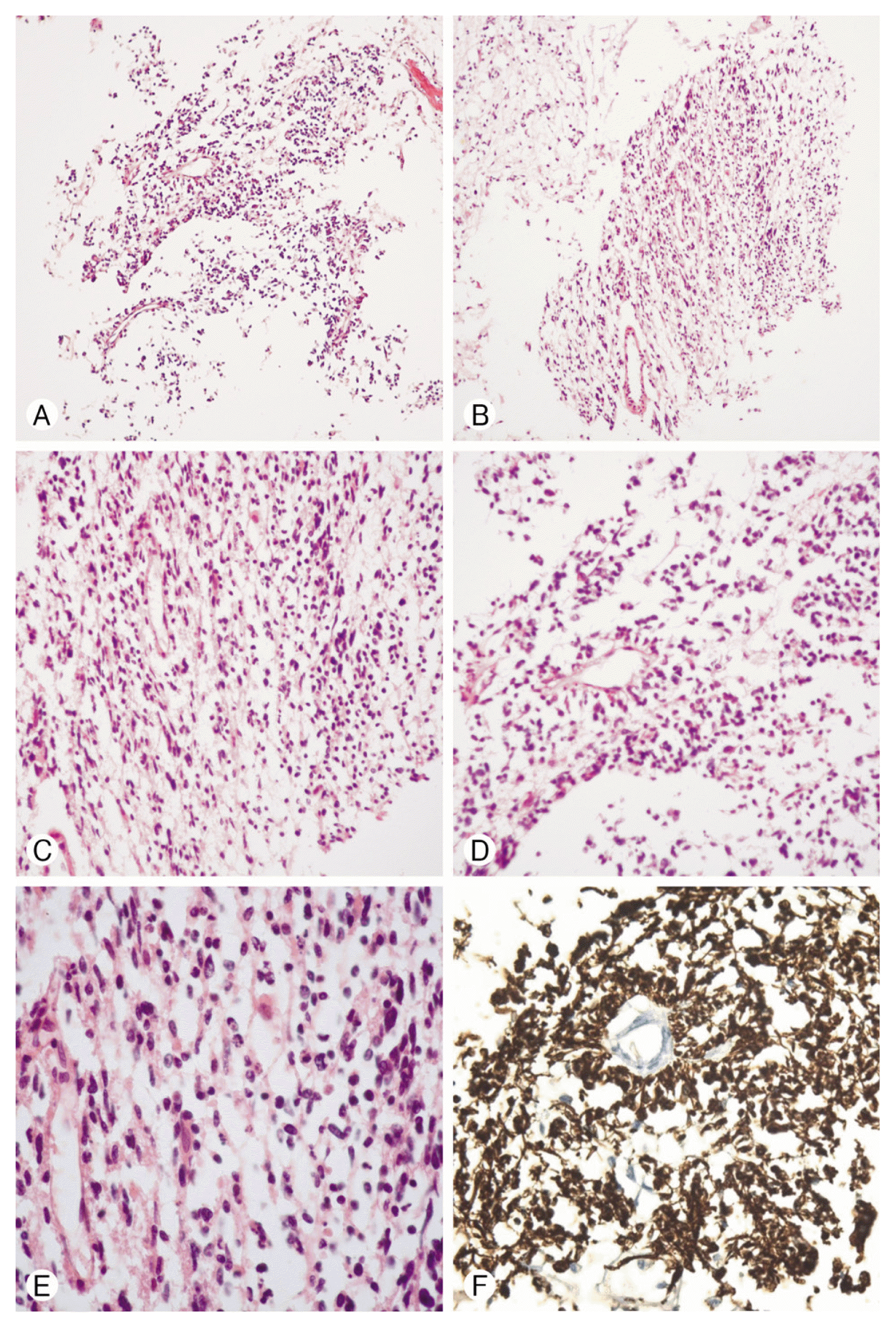
(A, B) Histological appearance of areas of tumor, Hema-toxyline-Eosin (HE) staining (magnification ×100): shows the microvascular proliferation of astrocytic tumor cells. Perivascular tumor cells which are mostly comprised of small undifferentiated cells. Also, microscopic findings show evidence of geographical pseudo-palisading tumor necrosis. The presence of either of these factors in an astrocytic tumor is sufficient to make a diagnosis of glioblastoma. (C, D) HE staining (magnification ×200) illustrating small round blue cells with high mitotic rates and pleomorphic nuclei characteristic of a PNET. (E) HE staining (magnification ×400) shows highly cellular neoplasia with hyperchromatic pleomorphic nuclei and mitosis. (F) Immunohistochemical features of the tumor (magnification ×200). Some tumor cells are immunoreactive for GFAP.
The patient was diagnosed with ‘glioblastoma with a PNET component’. After adjuvant radiotherapy of 6,300 cGY, there was no recurrence apparent on a follow-up MRI six months later (Fig. 2). This patient showed an alert mentality without neurologic deficit during the 12 months follow-up.

12 months later follow-up MRI after first operation and radiotherapy (Axial T1-WI MRI with enhancement): MRI obtained 12 months after surgery and conventional radiation therapy shows a decrease in the swelling of the portion of the masses, and shows no evidence of tumor recurrence. Images shows a decrease in the extent of the enhancing portion of the masses.
DISCUSSION
Glioblastoma, classified by World Health Organization (WHO) as a grade IV glioma, is the most frequent primary malignant tumor of the brain in adults. The prognosis is poor despite more recent advances in multi-modal treatment, which includes radiotherapy combined with chemotherapy, and uses pharmacological agents such as temozolomide10,12,17). The standard of treatment for patients with newly diagnosed glioblastoma includes surgical resection of the tumor followed by a 6-week course of radiotherapy with concomitant systemic therapy using an alkylating agent (temozolomide), which is then followed by the minimum of 6 months of adjuvant temozolomide. Most patients diagnosed with this tumor die within one year from the diagnosis and only 5% survive more than 5 years despite aggressive therapies17).
Some classifications have been studied to organize the heterogeneity of glioblastoma. Recently, the criteria designating a glioblastoma variant in comparison to the histopathological patterns of differentiation remain to be considered. The 2007 WHO guidelines for brain tumors have suggested the possibility of various emerging glioblastoma variants10). Central nervous system tumors with combined features of high-grade glioma and primitive neuroectodermal are rare and poorly characterized. Theoretically, astrocytic tumor cells should be positive for GFAP. Glioblastoma with a small cell component is a rare finding in neuronal immunophenotype. These ‘undifferentiated’ or ‘embryonal’ cells generally are not immunoreactive to GFAP4). In our case, the tumor consisted of two different components which were histologically distinct. Immunohistochemically, GFAP positivity was found in the astrocytic cells and fibrillary network. This supported the glial nature of the tumor. Another component of the tumor showed a multifocal distribution and consisted of cellular areas of small cells with narrow cytoplasms and hyperchromatic nuclei. The cells in these areas occasionally formed pseudorosettes and their mitotic activity was high. Recent reports have suggested the existence of ‘glioblastoma with PNET component’ that contains two distinct architectures, including that of traditional glioblastoma and that of PNET-like areas with hypercellularity, a minimal fibrillary background, small undifferentiated cells with scant cytoplasm, oval round hyperchromatic nuclei, and Hormer Wright neuroblastic rosettes, as a potential variant of glioblastoma found in the elderly6,7,14).
The most recent and largest report studied 53 cases and was performed by Perry et al.14) Although tumors with combined features of glioblastoma with PNET are rare, a motivating factor for the current study was the challenges they pose, both to the pathologist and to the oncologist. The tumors are encountered more commonly in adults and the occurrence of symptoms takes a considerably shorter time (less than 6 months). Most of tumors are localized in the temporal lobe, whereas they are rarely seen within infratentorial localization. Radiologically, intratumoral hemorrhage is observed in few of cases along the necrotic and cystic areas. The tumor demonstrates a significant mass effect and exhibits diffuse peritumoral edema. Having a well-circumscribed character facilitates the surgical excision. Perry et al. and Varlet et al. evaluated those types of tumors as a histological sub-group of glioblastoma and described them with the term ‘Glioblastoma with PNET’14,18). Rare examples of high-grade glioneuronal tumors or glioblastoma combined with PNET have been previously published but remain relatively poorly characterized.
In contrast, recent studies have suggested favorable prognostic features for glioblastoma with a PNET component, like in our case. A study of 40 cases of glioblastoma with PNET components demonstrated that the coexpression of GFAP and NFP (neurofilament protein) was observed with a low recurrence rate (36%) and, after surgery, a mean survival of 44 months18). In another study of 12 patients with glioblastoma with PNET, mutations in isocitrate dehydrogenase 1 were seen in 25% of cases evaluated with overall survival of 15 and 31 months, respectively16).
Compared with classic glioblastoma, these tumors exhibit a lower rate of local recurrence following total resection, while manifesting a more common metastatic spread. There are investigators who argue that local cranio-spinal radiation therapy as an inadequate method for preventing the tumor from spreading, and mention chemotherapy as a more efficient therapeutic treatment18). Standard protocols for radiotherapy and chemotherapy processes have not been identified yet, due to the rarity and poor prognosis of the disease. In our case, tumor removal and adjuvant radiotherapy with a total dose over 6,300 cGY were performed.
The patient’s age, tumor localization, and radiological findings are important to the clinical differential diagnosis. A definitive diagnosis of glioblastoma with PNET includes other types of glioneuronal tumors, embryonal tumors, and metastatic neuroendocrine carcinomas that require comprehensive histopathological and immunohistochemical examinations13).
CONCLUSION
We report a case of glioblastoma with a PNET component, a rare form of glioma, which has a relatively favorable outcome after surgery with radiotherapy.
ACKNOWLEDGEMENTS
This study was supported by BioGreen 21 program(PJ01121401) of Rural Development Administration.